Start here: your first disease progress workflow
Kaique S. Alves
2026-05-17
Source:vignettes/getting-started.Rmd
getting-started.Rmd
library(epifitter)
library(dplyr)
library(ggplot2)
library(cowplot)
theme_set(cowplot::theme_half_open(font_size = 12))What epifitter does
epifitter helps analyze plant disease progress curves
(DPCs): repeated measurements of disease intensity through time. These
curves are common in plant pathology because they connect epidemic
development, host-pathogen biology, management effects, and quantitative
summaries such as apparent infection rate and area under the disease
progress curve.
This article is the shortest path to a complete workflow. You will:
- simulate a synthetic epidemic;
- fit canonical disease progress models;
- inspect fitted parameters and model rankings;
- plot observed and fitted disease progress curves;
- see how the same workflow extends to multiple epidemics.
The examples use simulated data so that the workflow is reproducible, but the same functions can be applied to observed disease incidence or severity data on a proportional scale.
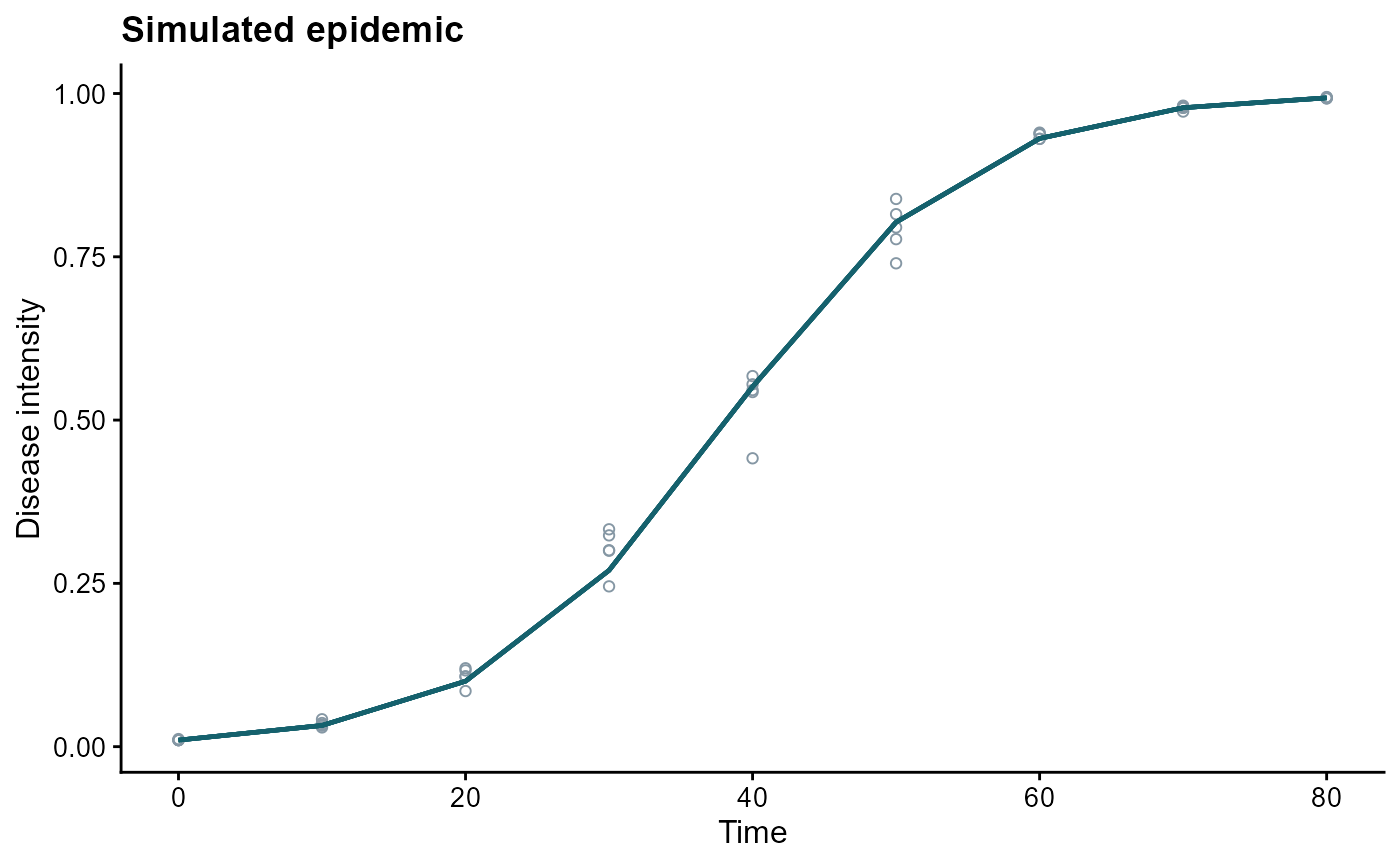
A first epidemic
We start by simulating a logistic epidemic. The column y
is the underlying curve and random_y represents replicated
observations with sampling or measurement noise.
set.seed(1)
epi <- sim_logistic(
N = 80,
y0 = 0.01,
dt = 10,
r = 0.12,
alpha = 0.2,
n = 5
)
knitr::kable(head(epi), digits = 4)| replicates | time | y | random_y |
|---|---|---|---|
| 1 | 0 | 0.0100 | 0.0100 |
| 1 | 10 | 0.0325 | 0.0336 |
| 1 | 20 | 0.1002 | 0.0851 |
| 1 | 30 | 0.2699 | 0.3328 |
| 1 | 40 | 0.5511 | 0.5674 |
| 1 | 50 | 0.8030 | 0.7770 |
ggplot(epi, aes(time, y, group = replicates)) +
geom_point(aes(y = random_y), shape = 1, color = "#8597a4") +
geom_line(color = "#15616d", linewidth = 0.8) +
labs(
title = "Simulated epidemic",
x = "Time",
y = "Disease intensity"
)
Fit candidate models
fit_lin() compares the four canonical disease progress
models by fitting linearized forms of each model. This is useful for
quick screening and teaching, but the ranking should be interpreted
together with the plot and biological plausibility.
fit <- fit_lin(time = epi$time, y = epi$random_y)
fit## Results of fitting population models
##
## Stats:
## CCC r_squared RSE
## Logistic 0.9988 0.9976 0.1561
## Gompertz 0.9774 0.9558 0.4734
## Monomolecular 0.9313 0.8714 0.6420
## Exponential 0.9161 0.8452 0.6337
##
## Infection rate:
## Estimate Std.error Lower Upper
## Logistic 0.11931599 0.0009014682 0.11749801 0.12113397
## Gompertz 0.08329917 0.0027332800 0.07778699 0.08881135
## Monomolecular 0.06325781 0.0037064427 0.05578306 0.07073257
## Exponential 0.05605817 0.0036589168 0.04867927 0.06343708
##
## Initial inoculum:
## Estimate Linearized lin.SE Lower Upper
## Logistic 1.058484e-02 -4.5376913 0.04291847 9.715742e-03 0.011530776
## Gompertz 7.809693e-05 -2.2468144 0.13013015 4.571513e-06 0.000692952
## Monomolecular -1.515280e+00 -0.9223841 0.17646197 -2.590364e+00 -0.762114625
## Exponential 2.690866e-02 -3.6153072 0.17419928 1.893746e-02 0.038235118fit$stats_all contains the full ranking of candidate
models.
knitr::kable(fit$stats_all, digits = 4)| best_model | model | r | r_se | r_ci_lwr | r_ci_upr | v0 | v0_se | r_squared | RSE | CCC | y0 | y0_ci_lwr | y0_ci_upr |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Logistic | 0.1193 | 0.0009 | 0.1175 | 0.1211 | -4.5377 | 0.0429 | 0.9976 | 0.1561 | 0.9988 | 0.0106 | 0.0097 | 0.0115 |
| 2 | Gompertz | 0.0833 | 0.0027 | 0.0778 | 0.0888 | -2.2468 | 0.1301 | 0.9558 | 0.4734 | 0.9774 | 0.0001 | 0.0000 | 0.0007 |
| 3 | Monomolecular | 0.0633 | 0.0037 | 0.0558 | 0.0707 | -0.9224 | 0.1765 | 0.8714 | 0.6420 | 0.9313 | -1.5153 | -2.5904 | -0.7621 |
| 4 | Exponential | 0.0561 | 0.0037 | 0.0487 | 0.0634 | -3.6153 | 0.1742 | 0.8452 | 0.6337 | 0.9161 | 0.0269 | 0.0189 | 0.0382 |
Visualize predictions
The fitted object stores observed values, transformed values,
predictions, and parameter summaries. plot_fit() turns that
object into a faceted ggplot2 figure.
plot_fit(fit, point_size = 1.8, line_size = 0.9)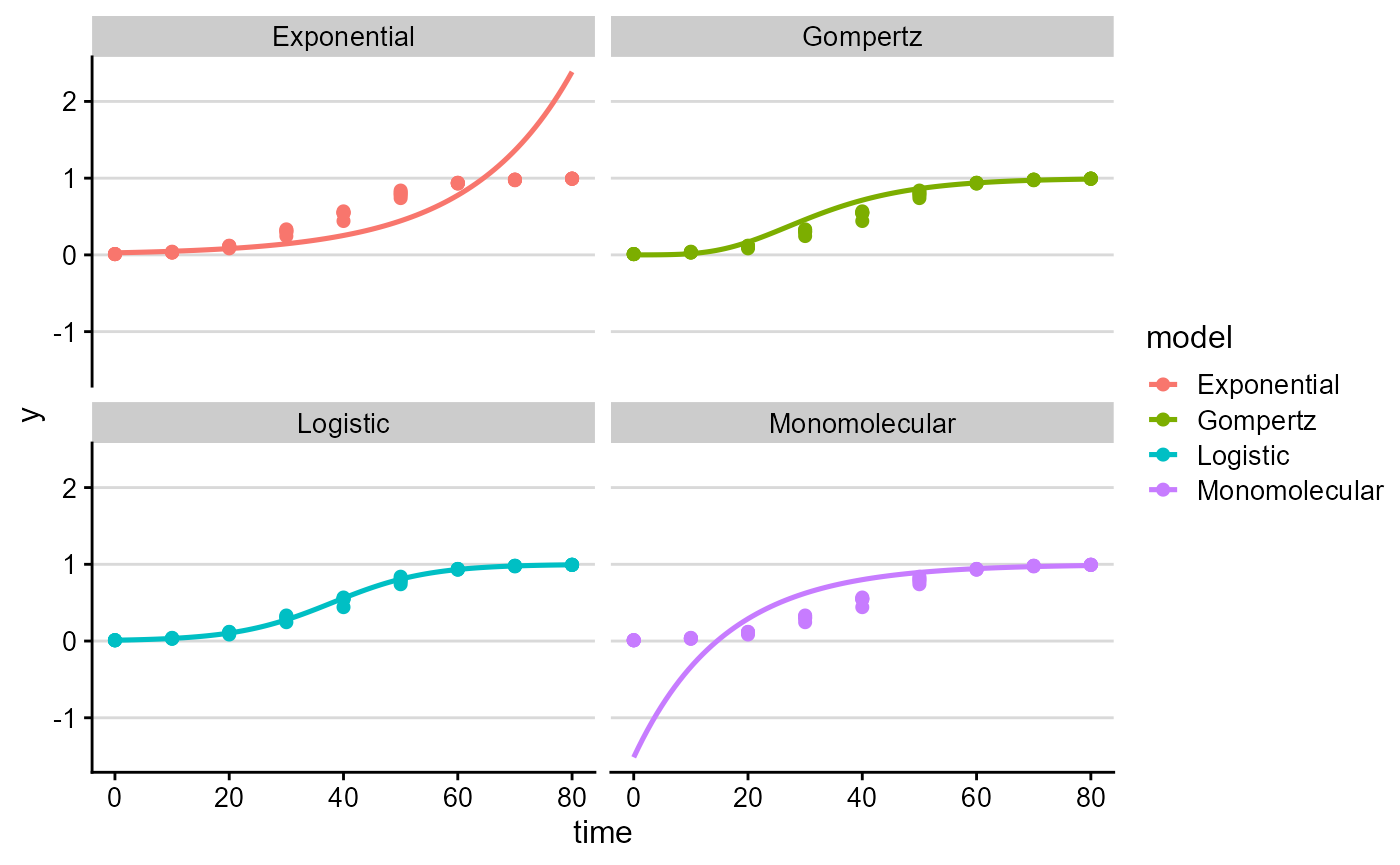
Work with multiple epidemics
Many experiments include multiple treatments, cultivars, blocks, or
locations. Use fit_multi() when the same fitting workflow
should be applied separately to each group.
epi_a <- sim_gompertz(N = 50, y0 = 0.002, dt = 5, r = 0.10, alpha = 0.2, n = 3)
epi_b <- sim_gompertz(N = 50, y0 = 0.002, dt = 5, r = 0.14, alpha = 0.2, n = 3)
multi_epi <- bind_rows(epi_a, epi_b, .id = "epidemic")
multi_fit <- fit_multi(
time_col = "time",
intensity_col = "random_y",
data = multi_epi,
strata_cols = "epidemic"
)
knitr::kable(head(multi_fit$Parameters), digits = 4)| epidemic | best_model | model | r | r_se | r_ci_lwr | r_ci_upr | v0 | v0_se | r_squared | RSE | CCC | y0 | y0_ci_lwr | y0_ci_upr |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | Gompertz | 0.0997 | 0.0017 | 0.0962 | 0.1032 | -1.7976 | 0.0513 | 0.9907 | 0.1575 | 0.9953 | 0.0024 | 0.0012 | 0.0044 |
| 1 | 2 | Monomolecular | 0.0671 | 0.0032 | 0.0605 | 0.0737 | -0.4718 | 0.0957 | 0.9327 | 0.2939 | 0.9652 | -0.6028 | -0.9484 | -0.3186 |
| 1 | 3 | Logistic | 0.1655 | 0.0090 | 0.1473 | 0.1838 | -4.3343 | 0.2650 | 0.9168 | 0.8136 | 0.9566 | 0.0129 | 0.0076 | 0.0220 |
| 1 | 4 | Exponential | 0.0984 | 0.0115 | 0.0751 | 0.1218 | -3.8625 | 0.3390 | 0.7042 | 1.0408 | 0.8264 | 0.0210 | 0.0105 | 0.0420 |
| 2 | 1 | Gompertz | 0.1404 | 0.0018 | 0.1367 | 0.1441 | -1.7956 | 0.0537 | 0.9949 | 0.1648 | 0.9974 | 0.0024 | 0.0012 | 0.0045 |
| 2 | 2 | Monomolecular | 0.1102 | 0.0036 | 0.1028 | 0.1175 | -0.6420 | 0.1069 | 0.9677 | 0.3282 | 0.9836 | -0.9003 | -1.3632 | -0.5281 |
Next steps
- Use the PowderyMildew article for a real-data workflow based on the bundled experimental dataset.
- Use the model fitting article for a deeper walkthrough of
fit_lin(),fit_nlin(),fit_nlin2(), weighted nonlinear fits, andfit_multi(). - Use the confidence interval article when you need uncertainty bands around fitted curves.
- Use the area-under-the-curve article when your goal is to summarize each epidemic as AUDPC or AUDPS.
- Use the simulation workflow article for examples built around the
sim_family.